Research in the UMass MRSEC is organized into Interdisciplinary Research Groups (IRGs), each consisting of teams of researchers engaged in interdisciplinary projects.
Highlights
Probing Polymer Structure with Light
Engineering specific interactions in polymers, such as “face-to-face” or “head-to-tail” interactions, is central to advancing polymer-based optoelectronics. Barnes, Hayward and Emrick, working collaboratively in the Materials Research Science and Engineering Center on Polymers at UMass, showed that crystalline 2-D “nanowires” of conjugated polymers exhibit both types of interactions. Using single-molecule spectroscopy techniques, isolated nanowires were imaged and probed spectroscopically to reveal distinct spectral signatures of both J-type (head-to-tail), and H-type (face-to-face) electronic interactions. Controlling the directionality of such interactions is important for directing charge- and energy flow in solar cells and related devices.
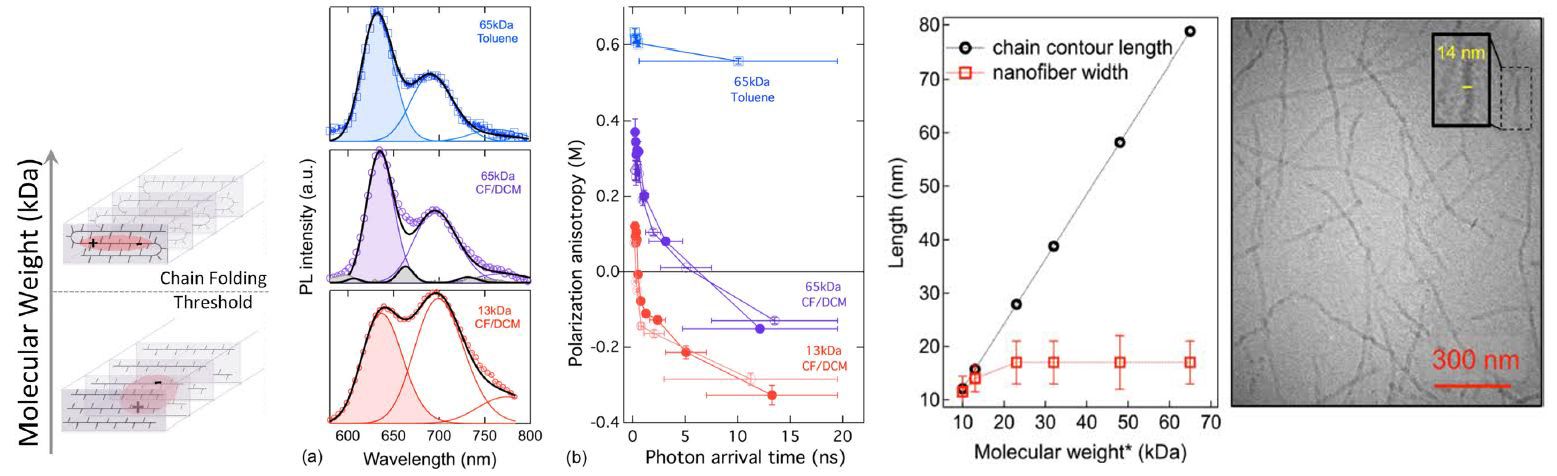
Polymer Molding and Self-Healing
Polymer materials exhibit numerous useful characteristics at multiple length scales, from the angstrom scale (atom-to-atom bonding) to the nanoscale (interactions of groups of atoms) to the macroscale (collective properties of large objects). McCarthy, working in the Materials Reseach Science and Engineering Center on Polymers at UMass, showed that silicon-containing polymers, termed siloxanes, undergo self-healing events that allow a variety of shaping and re-shaping events, such as the “dog bone” to “dog” transition shown in the picture. Importantly, pristine mechanical properties are maintained in all of the shapes, due to the ability of the siloxane bonding structure to heal during processing.
Perfectly Organized Gold Nanorings
 Creating well-organized conducting nanostructures in a flexible polymer matrix provides platforms for numerous applications in optics, sensors, and wave-guiding structures. Working in the Materials Research Science and Engineering Center on Polymers at the University of Massachusetts Amherst, Thayumanavan and Russell achieved self-assembled hybrid structures from diblock copolymers and gold nanoparticles, where the nanoparticles are exquisitely arranged in circular patterns as directed by the polymer template. Key to this process was the synthesis of block copolymers having a photocleavable junction point; after light-initiated polymer fragmentation, the template is left with sulfur-rich functional groups having an affinity for metallic surfaces, such as gold and silver nanoparticles. The accompanying image shows the results of this process, with the dark circular areas indicating the presence of gold nanoparticles arranged along the edges of the circular polymer pattern.
Creating well-organized conducting nanostructures in a flexible polymer matrix provides platforms for numerous applications in optics, sensors, and wave-guiding structures. Working in the Materials Research Science and Engineering Center on Polymers at the University of Massachusetts Amherst, Thayumanavan and Russell achieved self-assembled hybrid structures from diblock copolymers and gold nanoparticles, where the nanoparticles are exquisitely arranged in circular patterns as directed by the polymer template. Key to this process was the synthesis of block copolymers having a photocleavable junction point; after light-initiated polymer fragmentation, the template is left with sulfur-rich functional groups having an affinity for metallic surfaces, such as gold and silver nanoparticles. The accompanying image shows the results of this process, with the dark circular areas indicating the presence of gold nanoparticles arranged along the edges of the circular polymer pattern.
A Simple Approach to Chiral Block Copolymer Assemblies
 Helical nanostructures via self-assembly are of interest for a variety of applications in the life and physical sciences. To date efforts to realize helical superstructures in block copolymers have focused on the use of a chiral monomers to incorporate chiral blocks directly into the main chain. In IRG 1, Watkins has demonstrated a simpler and more versatile approach that employs a small molecule chiral additive to drive the formation of helical morphologies in otherwise achiral block copolymer systems
Helical nanostructures via self-assembly are of interest for a variety of applications in the life and physical sciences. To date efforts to realize helical superstructures in block copolymers have focused on the use of a chiral monomers to incorporate chiral blocks directly into the main chain. In IRG 1, Watkins has demonstrated a simpler and more versatile approach that employs a small molecule chiral additive to drive the formation of helical morphologies in otherwise achiral block copolymer systems
Homogenizing Inhomogeneous Wrinkles
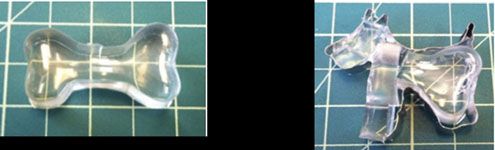
 Understanding the commonplace mechanical instability known as “wrinkling” has the potential to impact numerous materials and technological applications, such as optical surfaces, adhesives and flexible electronic devices. Understanding how wrinkles change with strain is important for many of these applications. While homogeneous strain will result in a homogeneously wrinkled surface, a systematic understanding of pre-existing strain inhomogeneity, which is commonly found in practical materials, has remained elusive. Crosby, leading a collaborative effort in the Materials Research Science and Engineering Center on Polymers at UMass, developed novel fabrication methods to find that a surface with inhomogeneously distributed wrinkles will develop a surprising pattern of wrinkles on the surface, in which the wrinkles have exhibit homogeneous geometric dimensions at very small applied strains (~6%).
Understanding the commonplace mechanical instability known as “wrinkling” has the potential to impact numerous materials and technological applications, such as optical surfaces, adhesives and flexible electronic devices. Understanding how wrinkles change with strain is important for many of these applications. While homogeneous strain will result in a homogeneously wrinkled surface, a systematic understanding of pre-existing strain inhomogeneity, which is commonly found in practical materials, has remained elusive. Crosby, leading a collaborative effort in the Materials Research Science and Engineering Center on Polymers at UMass, developed novel fabrication methods to find that a surface with inhomogeneously distributed wrinkles will develop a surprising pattern of wrinkles on the surface, in which the wrinkles have exhibit homogeneous geometric dimensions at very small applied strains (~6%).
Creating a "repair-and-go" system using nanoparticle microencapsulation
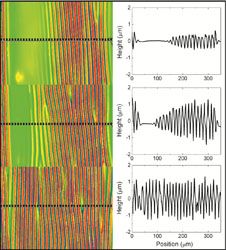
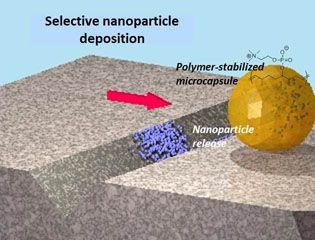 Facile methods for detecting and repairing damaged regions of materials are critically important in numerous structural and functional materials, from airplane wings to fabrics to microelectronics to biological implant materials. Emrick, Crosby, and Russell, working in the Materials Research Science and Engineering Center (MRSEC) on Polymers at UMass Amherst, demonstrated that microcapsules can carry nanoparticles across a damaged substrate, sense the damaged regions, and deposit nanoparticles selectively into the damaged areas, leaving the rest of the surface unaffected. The work represents an experimental realization of a theoretical challenge described by Balazs (U Pittsburgh), termed “repair-and-go”. The success of “repair-and-go” hinges on microencapsulation with polymer surfactants, as the thin polymer capsule wall (the polymer structure of the capsules is shown in the figure) allows nanoparticles to pass out of the capsule and into the crack. Moreover, the polymer is anti-fouling, preventing irreversible absorption of the capsule on the substrate or in the damaged region.
Facile methods for detecting and repairing damaged regions of materials are critically important in numerous structural and functional materials, from airplane wings to fabrics to microelectronics to biological implant materials. Emrick, Crosby, and Russell, working in the Materials Research Science and Engineering Center (MRSEC) on Polymers at UMass Amherst, demonstrated that microcapsules can carry nanoparticles across a damaged substrate, sense the damaged regions, and deposit nanoparticles selectively into the damaged areas, leaving the rest of the surface unaffected. The work represents an experimental realization of a theoretical challenge described by Balazs (U Pittsburgh), termed “repair-and-go”. The success of “repair-and-go” hinges on microencapsulation with polymer surfactants, as the thin polymer capsule wall (the polymer structure of the capsules is shown in the figure) allows nanoparticles to pass out of the capsule and into the crack. Moreover, the polymer is anti-fouling, preventing irreversible absorption of the capsule on the substrate or in the damaged region.
Conjugated polymer helices
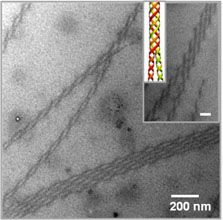 Polymers having “conjugated structures”, allowing them to conduct electricity, hold great potential for flexible and ink-jet printable electronic devices and inexpensive plastic solar cells. Work by Hayward and Emrick in the Materials Research Science and Engineering Center (MRSEC) on Polymers at the University of Massachusetts Amherst has shown how to coax such polymers to twist into conducting wires thousands of times smaller than the twisted cables used in common electronic devices. These structures are based on polythiophenes, which tend to crystallize into ribbons only a few nanometers in thickness. Such crystalline nanostructures are important to determining the electronic properties of materials based on this polymer, and the ability to control their formation holds promise for improving the performance of devices. Remarkably, the inclusion of functional groups that bind salt ions leads to twisting of these nanowires into helices that join together into double helices, reminiscent of DNA, and larger bundles containing multiple strands. These materials provide opportunities to 1) study the still poorly understood driving forces for helical assembly, and 2) tune the electronic properties of conjugated polymers.
Polymers having “conjugated structures”, allowing them to conduct electricity, hold great potential for flexible and ink-jet printable electronic devices and inexpensive plastic solar cells. Work by Hayward and Emrick in the Materials Research Science and Engineering Center (MRSEC) on Polymers at the University of Massachusetts Amherst has shown how to coax such polymers to twist into conducting wires thousands of times smaller than the twisted cables used in common electronic devices. These structures are based on polythiophenes, which tend to crystallize into ribbons only a few nanometers in thickness. Such crystalline nanostructures are important to determining the electronic properties of materials based on this polymer, and the ability to control their formation holds promise for improving the performance of devices. Remarkably, the inclusion of functional groups that bind salt ions leads to twisting of these nanowires into helices that join together into double helices, reminiscent of DNA, and larger bundles containing multiple strands. These materials provide opportunities to 1) study the still poorly understood driving forces for helical assembly, and 2) tune the electronic properties of conjugated polymers.
Mesoscopically helical order in chiral block copolymers
 Supermolecular, helical assemblies are a common structural motif exploited in far-ranging biological contexts, from the flagellar appendages of swimming microorganisms to the protein coats that sheath rod-like viruses. The screw-like structure of these biological assemblies is a consequence of the chiral subunits (proteins and amino acids) from which they are built, and this chirality imbues the structures with a right- or left-handed sense or twist. Grason and Russell, working collaboratively at the Materials Research Science and Engineering Center at UMass Amherst, demonstrate that a similar strategy may be employed in synthetic polymers, which are seen to self-assemble into helical structures. In general, block copolymers are assemble into array of mirror-symmetric, nano-structured arrays of ordered spherical, cylindrical and layered domains. This work uncovered a novel theoretical tool to both model and predict the self-assembled structures that emanate from constructing one of the constituent polymeric blocks from chiral segments. The theory shows that interactions between chiral segments “twist” the trajectories of the self-organized chain molecules. This twist is transmitted through the tension carried along the chains to the entire assembly, driving otherwise cylindrical assemblies to “buckle” into nanohelices organized in a hexagonal array. These results confirm the notion that mesoscopic, helical structures observed on tens of nanometer length scales in melts of the chiral block copolymer, poly-L-lactide-b-polystyrene, stems from the chiral nature of inter-molecular forces taking place at the nanometer lengthscale. Going forward, this opens a door to a new class of complex meso-chiral structures formed in chiral block copolymer materials yet to be discovered.
Supermolecular, helical assemblies are a common structural motif exploited in far-ranging biological contexts, from the flagellar appendages of swimming microorganisms to the protein coats that sheath rod-like viruses. The screw-like structure of these biological assemblies is a consequence of the chiral subunits (proteins and amino acids) from which they are built, and this chirality imbues the structures with a right- or left-handed sense or twist. Grason and Russell, working collaboratively at the Materials Research Science and Engineering Center at UMass Amherst, demonstrate that a similar strategy may be employed in synthetic polymers, which are seen to self-assemble into helical structures. In general, block copolymers are assemble into array of mirror-symmetric, nano-structured arrays of ordered spherical, cylindrical and layered domains. This work uncovered a novel theoretical tool to both model and predict the self-assembled structures that emanate from constructing one of the constituent polymeric blocks from chiral segments. The theory shows that interactions between chiral segments “twist” the trajectories of the self-organized chain molecules. This twist is transmitted through the tension carried along the chains to the entire assembly, driving otherwise cylindrical assemblies to “buckle” into nanohelices organized in a hexagonal array. These results confirm the notion that mesoscopic, helical structures observed on tens of nanometer length scales in melts of the chiral block copolymer, poly-L-lactide-b-polystyrene, stems from the chiral nature of inter-molecular forces taking place at the nanometer lengthscale. Going forward, this opens a door to a new class of complex meso-chiral structures formed in chiral block copolymer materials yet to be discovered.
Inside a crumpled ball
 The crumpling or crushing of paper, aluminum foil, or even a car fender is an everyday occurrence that is surprisingly rich in new physical and materials principles. Working in the Materials Research Science and Engineering Center (MRSEC) on Polymers at the University of Massachusetts Amherst, Menon and Russell used X-ray microtomography experiments on foils crushed into a ball to understand their detailed 3D structure. These measurements reveal that the internal 3-dimensional geometry of a crumpled ball is in many respects isotropic and homogeneous. Crumpling recapitulates classic nonequilibrium problems such as turbulence, where a system driven by long-wavelength and low-symmetry forces shows only rather subtle fingerprints of the forcing mechanism. However, the researchers found local nematic ordering of the sheet into parallel stacks, a surprising results that will help physicists and materials scientists better understand these complex structures. The extent of this stacking or layering increased with the volume fraction, or degree of compression. Stacking of the material into thicker “walls” may be either an alternative or an assistive mechanism to the formation of ridges, and impart structural rigidity to the material.
The crumpling or crushing of paper, aluminum foil, or even a car fender is an everyday occurrence that is surprisingly rich in new physical and materials principles. Working in the Materials Research Science and Engineering Center (MRSEC) on Polymers at the University of Massachusetts Amherst, Menon and Russell used X-ray microtomography experiments on foils crushed into a ball to understand their detailed 3D structure. These measurements reveal that the internal 3-dimensional geometry of a crumpled ball is in many respects isotropic and homogeneous. Crumpling recapitulates classic nonequilibrium problems such as turbulence, where a system driven by long-wavelength and low-symmetry forces shows only rather subtle fingerprints of the forcing mechanism. However, the researchers found local nematic ordering of the sheet into parallel stacks, a surprising results that will help physicists and materials scientists better understand these complex structures. The extent of this stacking or layering increased with the volume fraction, or degree of compression. Stacking of the material into thicker “walls” may be either an alternative or an assistive mechanism to the formation of ridges, and impart structural rigidity to the material.
Responsive wrinkles
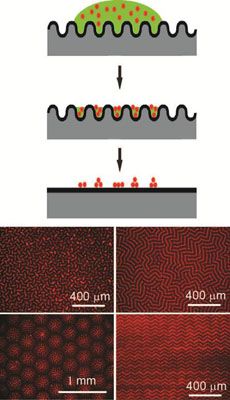 Wrinkling, buckling, and creasing phenomena in synthetic and natural materials represents an opportunity to guide the placement of nanostructures that enhance the optical, mechanical, and/or electronic properties of materials. Working in the Materials Research Science and Engineering Center on Polymers at UMass Amherst, Crosby discovered that reversible wrinkling can guide colloidal and nanoparticle assembly into well-ordered patterns across macroscopic length scales. Due to the sinusoidal shape of top-constrained, swelling-induced wrinkling, the deposition of fluorescent polystyrene (PS) colloids occurs selectively in the wrinkle patterns during solvent evaporation. After complete evaporation of solvent, these colloidal assemblies are embossed as the surface snaps back to the smooth state (top schematic). A key advantage of this process is the ability to tune the assembly pattern by the solubility of the dispersing solvent in the wrinkled substrate. Solvent choice controls the applied strain that causes wrinkling, and the extent of strain beyond the critical value dictates the specific morphology obtained (bottom schematic).
Wrinkling, buckling, and creasing phenomena in synthetic and natural materials represents an opportunity to guide the placement of nanostructures that enhance the optical, mechanical, and/or electronic properties of materials. Working in the Materials Research Science and Engineering Center on Polymers at UMass Amherst, Crosby discovered that reversible wrinkling can guide colloidal and nanoparticle assembly into well-ordered patterns across macroscopic length scales. Due to the sinusoidal shape of top-constrained, swelling-induced wrinkling, the deposition of fluorescent polystyrene (PS) colloids occurs selectively in the wrinkle patterns during solvent evaporation. After complete evaporation of solvent, these colloidal assemblies are embossed as the surface snaps back to the smooth state (top schematic). A key advantage of this process is the ability to tune the assembly pattern by the solubility of the dispersing solvent in the wrinkled substrate. Solvent choice controls the applied strain that causes wrinkling, and the extent of strain beyond the critical value dictates the specific morphology obtained (bottom schematic).
A smooth cascade of structure refinement in thin sheets
 How does one make a rippled sheet terminate at a straight edge? If the sheet is sufficiently thin, such as a piece of paper or fabric, then the obvious solution of stretching it out flat will induce large stresses near the edge, possibly even tearing the sheet apart. One solution to this problem is suggested by the series of ever-smaller sharp folds of fabric generated near a curtain rod. Menon and Russell, working at the UMass Materials Research Science and Engineering Center, studied wrinkling patterns on an ultrathin floating raft of polystyrene, and discovered a new mechanism by which nature resolves such a conflict. In the middle of the film, a competition between gravity (which prefers shallow, frequent ripples) and the energy cost of bending the film (which favors longer, higher folds) determines the height and frequency of the folds. Near the edge, however, surface tension forces the film to lie flat. This refinement of structure occurred not by a hierarchy of pleats, but by a smooth cascade of ever-smaller wrinkles emerging as the edge is approached. This kind of hierarchical geometry is explained theoretically by the action of the surface tension of water that tends to “iron out” sharp features in the sheet. Similar types of smooth cascades may appear in other problems in materials science where a patterned surface hits an edge that is incompatible with the pattern.
How does one make a rippled sheet terminate at a straight edge? If the sheet is sufficiently thin, such as a piece of paper or fabric, then the obvious solution of stretching it out flat will induce large stresses near the edge, possibly even tearing the sheet apart. One solution to this problem is suggested by the series of ever-smaller sharp folds of fabric generated near a curtain rod. Menon and Russell, working at the UMass Materials Research Science and Engineering Center, studied wrinkling patterns on an ultrathin floating raft of polystyrene, and discovered a new mechanism by which nature resolves such a conflict. In the middle of the film, a competition between gravity (which prefers shallow, frequent ripples) and the energy cost of bending the film (which favors longer, higher folds) determines the height and frequency of the folds. Near the edge, however, surface tension forces the film to lie flat. This refinement of structure occurred not by a hierarchy of pleats, but by a smooth cascade of ever-smaller wrinkles emerging as the edge is approached. This kind of hierarchical geometry is explained theoretically by the action of the surface tension of water that tends to “iron out” sharp features in the sheet. Similar types of smooth cascades may appear in other problems in materials science where a patterned surface hits an edge that is incompatible with the pattern.
Nanoscale Imaging of wet polymer materials
The molecular- and nano-scale structure of bulk materials can be imaged only by transmission electron microscopy (TEM), a technique that requires high vacuum (low pressure). Until now, this constraint made TEM incompatible with ‘wet’ materials, such as molecules dispersed in a liquid or gels swollen by a solvent. Working at the Materials Research Science and Engineering Center (MRSEC) on Polymers at UMass Amherst, Hoagland, McCarthy, and Russell successfully imaged the fine structure of wet polymeric materials prepared with ionic liquids. These non-volatile liquids dissolve or solvate an interesting spectrum of ‘soft’ polymer systems of fundamental and technological interest. A key objective is to refine the approach so that the dynamics of such systems can be imaged in situ and in real time, accomplishing tasks akin to those known from optical microscopy but at much smaller length scales. Shown below are two images, taken 1 second apart, of an ionic liquid spanning a 1-mm opening in a carbon film. Suspended in the liquid are polymer-coated nanoparticle tracers. In the interval between images, the thin film, originally spread over the opening, has destabilized by breaking into ~100-nm droplets that distribute around the rim. Nano- and micro-scale wetting transitions are key to numerous technologies ranging from microfluidics to ultralyophobic surfaces, but these transitions have never been visualized experimentally.

Ionic liquids as solution media for bioconjugation
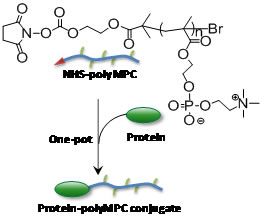 Water soluble polymers, once reserved for commodity applications (i.e., shaving cream, emulsification processes, etc.) have emerged as valuable materials for medicine. Combining synthetic polymers with therapeutic proteins and cancer drugs improves the “therapeutic index” of the drugs, preventing their fast elimination from the body, and improving their availability for treating the disease. Emrick at the UMass Materials Research Science and Engineering Center found that ionic liquids provide an excellent environment for the attachment of proteins to synthetic polymers. Water soluble polymers like “polyMPC” shown in the figure are of interest for conjugation to proteins for medicine. However, the conjugation reaction itself is problematic when performed in water, due to loss of the reactive end-group to hydrolysis. Ionic liquids were found to provide an alternative medium for this conjugation, as the polar but aqueous-free characteristics of ionic liquids prevent competition of water for the reactive site at the polymer-chain-end, leading to clean, efficient conjugation. Such efficient and convenient bioconjugation strategies are critically important for scale-up and optimization of new polymer-based conjugates for medicine.meso-chiral structures formed in chiral block copolymer materials yet to be discovered.
Water soluble polymers, once reserved for commodity applications (i.e., shaving cream, emulsification processes, etc.) have emerged as valuable materials for medicine. Combining synthetic polymers with therapeutic proteins and cancer drugs improves the “therapeutic index” of the drugs, preventing their fast elimination from the body, and improving their availability for treating the disease. Emrick at the UMass Materials Research Science and Engineering Center found that ionic liquids provide an excellent environment for the attachment of proteins to synthetic polymers. Water soluble polymers like “polyMPC” shown in the figure are of interest for conjugation to proteins for medicine. However, the conjugation reaction itself is problematic when performed in water, due to loss of the reactive end-group to hydrolysis. Ionic liquids were found to provide an alternative medium for this conjugation, as the polar but aqueous-free characteristics of ionic liquids prevent competition of water for the reactive site at the polymer-chain-end, leading to clean, efficient conjugation. Such efficient and convenient bioconjugation strategies are critically important for scale-up and optimization of new polymer-based conjugates for medicine.meso-chiral structures formed in chiral block copolymer materials yet to be discovered.
Surfactant-mediated ion exchange and charge reversal at ionic liquid interfaces
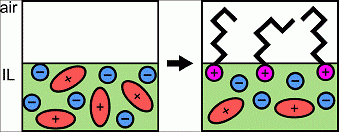 Bermudez at the UMass Materials Research Science and Engineering Center found that room-temperature ionic liquids (ILs) exhibit a unique set of properties due to their charged character, presenting opportunities for applications not possible using conventional organic solvents or water. This work showed combining positively or negatively charged molecules with ILs resulted in previously unknown interfacial behavior due to the electrostatic interactions between the charged molecule and the liquid. Specifically, sodium alkyl sulfates (negatively charged surfactants) and alkyl trimethylammonium bromides (positively charged surfactants) were found to segregate to the air-IL interface, and x-ray photoelectron spectroscopy revealed that the surfactant counter ions readily dissociate into the bulk, rather than staying close-by their oppositely charged partner. In addition, for an IL with an initial negative surface charge, the charge could be switched to positive by the addition of alkyl trimethylammonium bromides, giving access to a new method of manipulating surface charge of materials, which could have striking consequence as applied to coatings, ultra-thin films, and anti-fouling surfaces.
Bermudez at the UMass Materials Research Science and Engineering Center found that room-temperature ionic liquids (ILs) exhibit a unique set of properties due to their charged character, presenting opportunities for applications not possible using conventional organic solvents or water. This work showed combining positively or negatively charged molecules with ILs resulted in previously unknown interfacial behavior due to the electrostatic interactions between the charged molecule and the liquid. Specifically, sodium alkyl sulfates (negatively charged surfactants) and alkyl trimethylammonium bromides (positively charged surfactants) were found to segregate to the air-IL interface, and x-ray photoelectron spectroscopy revealed that the surfactant counter ions readily dissociate into the bulk, rather than staying close-by their oppositely charged partner. In addition, for an IL with an initial negative surface charge, the charge could be switched to positive by the addition of alkyl trimethylammonium bromides, giving access to a new method of manipulating surface charge of materials, which could have striking consequence as applied to coatings, ultra-thin films, and anti-fouling surfaces.


